
Schwerpunkt: Schwierigkeiten der Farbwahrnehmung
Genetisch bedingte oder erworbene Einschränkung?

Ein vollkommener Ausfall des Farbensehens, genannt Achromatopsie oder eine Einschränkung des Farbensehens, genannt Dyschromatopsie… Diese beiden Phänomene werden im Allgemeinen durch eine erblich bedingte Veränderungen der Zapfenzellen der Netzhaut hervorgerufen, können aber auch Folge eines Unfalls, einer Krankheit oder des Alterungsprozesses sein.
Von Dr. med. François-Xavier Borruat, Titularprofessor, Neuro-Ophthalmologie, Augenklinik Jules-Gonin, Lausanne und Dr. med. François Thommen, Oberarzt, Augenklinik Jules-Gonin, Lausanne
Ist eine Person von Achromatopsie betroffen, können keine Farben erkannt werden. Farbensichtigkeit setzt intakte Sehbahnen vom Auge bis ins Gehirn voraus. Im Bereich des Auges kommen beim Farbensehen in einem ersten Schritt die Zapfenzellen in der Netzhaut zum Einsatz. Dabei handelt es sich um lichtempfindliche Fotorezeptoren. Eine normale Farbensichtigkeit hängt somit zuerst vom Funktionieren der drei Typen von Zapfen im menschlichen Auge ab (S-Zapfen, M-Zapfen und L-Zapfen). Danach werden die visuellen Informationen entlang des Sehnervs über die Sehbahnen im Gehirn bis zu den Okzipitallappen geleitet, die ausschliesslich für die Verarbeitung von visuellen Reizen zuständig sind. Im Gehirn ist das visuelle Kortexareal V4 im parieto-okzipitalen Übergang auf die Farberkennung spezialisiert. Es gibt somit zwei mögliche Ursachen für eine Achromatopsie: Entweder ein Defekt der Zapfen-Fotorezeptoren der Netzhaut oder eine Läsion im hinteren Teil des Gehirns in Areal V4.
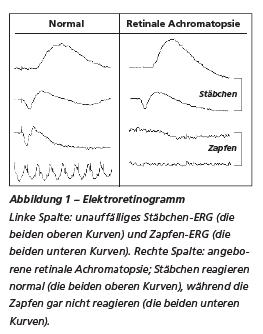
Retinale Achromatopsi1
Bei der retinalen Achromatopsie handelt es sich um eine meist autosomal-rezessive Erbkrankheit. Die Achromatopsie ist eine seltene Erkrankung mit einer Häufigkeitsverteilung von 1:30’000 bis 1:50’000 in der Bevölkerung. Das Krankheitsbild umfasst einen angeborenen Nystagmus (unkontrollierbare, rhythmisch verlaufende Bewegungen der Augen von links nach rechts oder umgekehrt), eine Photophobie (Überempfindlichkeit gegenüber Licht), eine schlechte Sehschärfe sowie eine stark eingeschränkte oder gar keine Farbwahrnehmung. Die Diagnose erfolgt über eine klinische Untersuchung sowie eine Bestätigung durch ein Elektroretinogramm (ERG; Messung der Elektroimpulse auf der Netzhaut, die aufgrund vorgegebener Lichtreize erzeugt werden), das eine fehlende elektrische Aktivität der Zapfen erkennen lässt, während die Stäbchen (spezialisierte Sinneszellen, die dem Nachtsehen dienen) ohne Einschränkung funktionieren (Abbildung 1). Ab einem Alter von fünf bis sechs Jahren kann auch eine multimodale Bildgebung erfolgen. Dabei werden eine Fotografie des Augenhintergrundes, Autofluoreszenz- und Pseudo-Infrarotaufnahmen der Netzhaut sowie eine optische Kohärenztomographie (OCT) durchgeführt.
Bis heute konnten Mutationen in sechs Genen (CNGA3, CNGB3, GNAT2, PDE6H, PDE6C, ATF6) als Ursache der retinalen Achromatopsie ermittelt werden. Bestimmte Mutationen scheinen sich nicht in einem besonderen klinischen Bild auszudrücken. Es ist somit nicht möglich, basierend auf der entdeckten Genmutation Aussagen über die Entwicklung des Sehvermögens zu machen. Lange Zeit betrachtete die Forschung die erblich bedingte retinale Achromatopsie als eine angeborene nicht fortschreitende Retinopathie. Aufgrund der jüngsten wissenschaftlichen Erkenntnisse ist man sich allerdings weitgehend einig, dass es sich bei der retinalen Achromatopsie um eine langsam fortschreitende Erkrankung handelt, bei der sich die Sehleistung mit der Zeit verschlechtert und auch die mittels OCT sichtbar gemachte Netzhautschädigung voranschreitet.
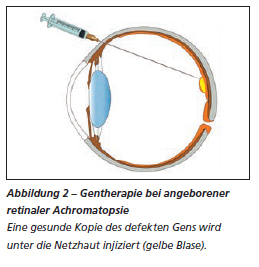
2007 ist es Forscherinnen und Forschern im Tierversuch erstmals gelungen, das Farbensehen bei Achromatopsie wiederherzustellen. Seither konnte in weiteren Studien die Wirksamkeit der Gentherapie in Tierversuchen (mit Mäusen, Hunden, Schafen, Affen) erfolgreich nachgewiesen werden. Bei Tieren wurde ein nachhaltiger Behandlungseffekt erzielt, wobei die Wirkung bei jüngeren Tieren stärker ist. Im Jahr 2017 wurden vier Studien zur Gentherapie beim Menschen lanciert (in Deutschland, England, Israel und den USA), bei denen nach Durchführung einer Vitrektomie (chirurgische Entfernung des Glaskörpers) Genmaterial über ein modifiziertes Virus unter die Netzhaut injiziert wird (Abbildung 2). Bis heute werden im Rahmen dieser Studien die Funktionen der am häufigsten mutierten Gene CNGA3 und CNGB3 ersetzt. Entsprechende Studienergebnisse werden zwischen 2020 und 2021 vorliegen.
Zerebrale Achromatopsie
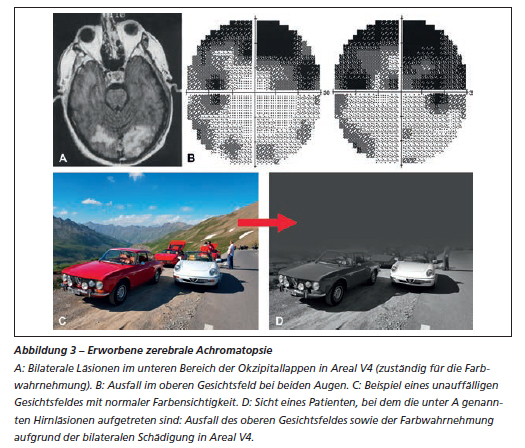
Bei der zerebralen Achromatopsie handelt es sich um eine äusserst seltene Folge eines Schlaganfalls. Im Allgemeinen kommt es zu einem sogenannten homonymen Gesichtsfeldausfall, bei dem das obere Gesichtsfeld betroffen ist. Es liegt kein Nystagmus vor, die Sehschärfe bleibt oft erhalten und der/die PatientIn zeigt keine Lichtscheu. Die Diagnose erfolgt über die Anamnese, eine klinische Untersuchung und mittels MRT, wobei eine Läsion im Bereich von Areal V4 festgestellt wird (Abbildung 4). Manchmal ist die zerebrale Achromatopsie auch eine dauerhafte Folge einer kortikalen Blindheit (Blindheit infolge einer Schädigung beider Okzipitallappen). Hier kehrt die anfänglich vollständig verlorene Sehfähigkeit langsam wieder zurück, allerdings sieht der Patient oder die Patientin nur noch schwarz-weiss und leidet unter einer Beeinträchtigung insbesondere des oberen Gesichtsfeldes. Für diese Patientinnen und Patienten gibt es bis heute keine Behandlungsmöglichkeiten.
Dyschromatopsie
Im Gegensatz zur Achromatopsie, bei der gar keine Farben mehr wahrgenommen werden, liegt bei der Dyschromatopsie entweder eine genetisch bedingte oder eine erworbene Farbenfehlsichtigkeit vor.
Bei den meisten erblich bedingten Farbenfehlsichtigkeiten besteht eine Anomalie bei einer (oder mehreren) Zapfenzellengruppe(n). Wie bereits erwähnt, gibt es drei verschiedene Zapfenarten (schematisch Rot-, Grün- und Blauzapfen). Zu den häufigsten Arten von Farbenfehlsichtigkeiten gehören die Protanopien und die Deuteranopien (Rot- bzw. Grünblindheit), die anomalen Trichromasien (Protanomalie und Deuteranomalie bzw. Rot- und Grünschwäche) und die S-Zapfen-Monochromasien (S-Zapfen, auch Blauzapfen genannt, sind empfindlich gegenüber kurzwelligem Licht), die infolge einer Funktionsstörung der Rot- und Grünzapfen auftreten. Diese Farbenfehlsichtigkeiten hängen mit rezessiven Mutationen auf dem X-Chromosom zusammen, weshalb Männer auch deutlich häufiger davon betroffen sind. Sie sind in der Regel nicht fortschreitend, und die betroffenen Personen verfügen ansonsten über ein ganz normales Sehvermögen.
Die genetisch bedingten Tritanopien (keine funktionierenden Blauzapfen) treten viel seltener auf und werden autosomal-dominant vererbt (über ein geschlechtsunabhängiges Chromosom). Ferner gehen sie mit einer verminderten Sehschärfe einher.
Darüber hinaus gibt es zahlreiche krankheitsbedingte Farbenfehlsichtigkeiten, die aufgrund von strukturellen/funktionellen Veränderungen der Zapfen, der Sehbahnen (insbesondere des Sehnervs) oder des primären visuellen Kortex und/oder dessen dazugehörigen assoziativen Arealen entstehen. Im Gegensatz zu den erblich bedingten Formen hat man es hier oft mit einer asymmetrischen und fortschreitenden Schädigung zu tun.
Die Farbwahrnehmung kann sich also mit fortschreitendem Alter verändern. Solche Veränderungen treten beispielsweise bei einer optischen Neuropathie oder bei Erkrankungen der Makula auf. Im Falle einer Trübung der optischen Medien (am häufigsten aufgrund des grauen Stars) kommt es zu einer spektralen Verschiebung beim sichtbaren Licht mit abnehmender Durchdringung kurzer Wellenlängen (blau/violett) und folglich zu einer Vergilbung des wahrgenommenen Bildes.